Pharmacology: Pharmacodynamics: Sildenafil is a potent and selective inhibitor of cyclic guanosine monophosphate (cGMP) specific phosphodiesterase type 5 (PDE5) the enzyme that is responsible for degradation of cGMP. Apart from the presence of this enzyme in the corpus cavernosum of the penis, PDE5 is also present in the pulmonary vasculature. Sildenafil, therefore, increases cGMP within pulmonary vascular smooth muscle cells resulting in relaxation. In patients with PAH this can lead to vasodilation of the pulmonary vascular bed and, to a lesser degree, vasodilatation in the systemic circulation.
Studies
in vitro have shown that sildenafil is selective for PDE5. Its effect is more potent on PDE5 than on other known phosphodiesterases. There is a 10-fold selectivity over PDE6 which is involved in the phototransduction pathway in the retina. There is an 80-fold selectivity over PDE1, and over 700-fold over PDE 2, 3, 4, 7, 8, 9, 10 and 11. In particular, sildenafil has greater than 4,000-fold selectivity for PDE5 over PDE3, the cAMP-specific phosphodiesterase isoform involved in the control of cardiac contractility.
Sildenafil causes mild and transient decreases in systemic blood pressure which, in the majority of cases, do not translate into clinical effects. The mean maximum decrease in supine systolic blood pressure following 100 mg oral dosing of sildenafil was 8.3 mmHg. The corresponding change in supine diastolic blood pressure was 5.3 mmHg.
After chronic dosing of 80 mg three times a day to healthy male volunteers, the largest average change from baseline of supine systolic blood pressure was a decrease of 9.0 mmHg. The corresponding change in supine diastolic blood pressure was a decrease of 8.4 mmHg.
After chronic dosing of 80 mg three times a day to patients with systemic hypertension the mean change from baseline in systolic and diastolic blood pressure was a decrease of 9.4 mmHg and 9.1 mmHg respectively.
After chronic dosing of 80 mg three times a day to patients with PAH lesser effects in blood pressure reduction were observed (a reduction in both systolic and diastolic pressure of 2 mmHg). This may be due to improvements in cardiac output secondary to the beneficial effects of sildenafil on pulmonary vascular resistance.
Single oral doses of sildenafil up to 100 mg in healthy volunteers produced no clinically relevant effects on ECG. After chronic dosing of 80 mg three times a day to patients with PAH no clinically relevant effects on the ECG were reported.
In a study of the hemodynamic effects of a single oral 100 mg dose of sildenafil in 14 patients with severe coronary artery disease (CAD) (>70% stenosis of at least one coronary artery), the mean resting systolic and diastolic blood pressures decreased by 7% and 6% respectively compared to baseline. Mean pulmonary systolic blood pressure decreased by 9%. Sildenafil showed no effect on cardiac output, and did not impair blood flow through the stenosed coronary arteries.
Mild and transient differences in color discrimination (blue/green) were detected in some subjects using the Farnsworth-Munsell 100 hue test at 1 hour following a 100 mg dose, with no effects evident after 2 hours post-dose. The postulated mechanism for this change in color discrimination is related to inhibition of PDE6, which is involved in the phototransduction cascade of the retina. Sildenafil has no effect on visual acuity, contrast sensitivity, electroretinograms, intraocular pressure, or pupillometry. In a small size placebo-controlled study of patients with documented early age-related macular degeneration (n=9), sildenafil (single dose, 100 mg) demonstrated no significant changes in visual tests conducted (visual acuity, Amsler grid, color discrimination simulated traffic light, Humphrey perimeter and photostress).
Efficacy in adult patients with PAH: A randomized, double-blind, placebo-controlled study was conducted in 278 patients with primary PAH, PAH associated with connective tissue disease (CTD), and PAH following surgical repair of congenital heart lesions. Patients were randomized to one of four treatment groups: placebo, sildenafil 20 mg, sildenafil 40 mg or sildenafil 80 mg, three times a day. Of the 278 patients randomized, 277 patients received at least 1 dose of study drug. The study population consisted of 68 (25%) men and 209 (75%) women with a mean age of 49 years (range: 18-81 years) and baseline 6-minute walk test distance (6MWD) between 100 and 450 meters (mean: 344 meters). 175 patients (63%) included were diagnosed with primary pulmonary hypertension, 84 (30%) were diagnosed with PAH associated with CTD and 18 (7%) of the patients were diagnosed with PAH following surgical repair of congenital heart lesions. Most patients were WHO Functional Class II (107/277, 39%) or III (160/277, 58%) with a mean baseline 6 minute walking distance of 378 meters and 326 meters respectively; fewer patients were Class I (1/277, 0.4%) or IV (9/277, 3%) at baseline. Patients with left ventricular ejection fraction <45% or left ventricular shortening fraction <0.2 were not studied.
Sildenafil (or placebo) was added to patients' background therapy, which could have included a combination of anticoagulants, digoxin, calcium channel blockers, diuretics and/or oxygen. The use of prostacyclin, prostacyclin analogues and endothelin receptor antagonists was not permitted neither was arginine supplementation. Patients who previously failed bosentan therapy were excluded from the study.
The primary efficacy endpoint was the change from baseline at Week 12 in 6MWD. A statistically significant increase in 6MWD was observed in all 3 sildenafil dose groups compared to those on placebo. Placebo corrected increases in 6MWD were 45 meters (p < 0.0001), 46 meters (p < 0.0001) and 50 meters (p < 0.0001) for sildenafil 20 mg, 40 mg and 80 mg respectively. There was no significant difference in effect between sildenafil doses.
The improvement in walk distance was apparent after 4 weeks of treatment and this effect was maintained at Weeks 8 and 12. Mean treatment effects consistently showed improvement in 6MWD in all sildenafil groups compared to placebo in all pre-defined subpopulations based on demographics, geographical regions, disease characteristics (in particular effects were similar among WHO functional class groups and etiologies) and baseline parameters (walk test and hemodynamics).
When analyzed by WHO functional class, a statistically significant increase in 6MWD was observed in the 20 mg dose group. For class II and class III, placebo corrected increases of 49 meters (p = 0.0007) and 45 meters (p = 0.0031) were observed respectively.
Patients on all sildenafil doses achieved a statistically significant reduction in mean pulmonary arterial pressure (mPAP) compared to those on placebo. Placebo-corrected treatment effects were -2.7 mmHg (p = 0.04), -3.0 mmHg (p = 0.01) and -5.1 mmHg (p < 0.0001) for sildenafil 20 mg, 40 mg and 80 mg respectively. Improvements were also seen in pulmonary vascular resistance (PVR), right atrial pressure (RAP) and cardiac output. Changes in heart rate and systemic blood pressure were negligible. The reduction in PVR was proportionally greater than the reduction in systemic vascular resistance (SVR). The incidence of clinical worsening events (in particular hospitalizations due to PAH) showed a favorable trend in the sildenafil treatment groups. A greater percentage of patients on each of the sildenafil doses (28%, 36% and 42% of subjects in sildenafil 20 mg, 40 mg and 80 mg, respectively) showed an improvement in at least 1 WHO functional class over the 12-week period compared to placebo (7%). Improvements were also seen in quality of life parameters, especially in physical functioning domains, and a favorable trend was seen Borg dyspnea score in sildenafil-treated patients compared to placebo. The percentage of subjects who had an addition of a class of background medication was greater in the placebo group (20%) compared to the active treatment groups (13% on sildenafil 20 mg; 16% on sildenafil 40 mg and 10% on sildenafil 80 mg).
Long-term Survival Data: Patients enrolled into the pivotal study were eligible to enter a long-term, open-label extension study. A total of 207 patients were treated with Revatio in the pivotal study, and their long-term survival status was assessed for a minimum of 3 years. In this population, Kaplan-Meier estimates of 1, 2 and 3 year survival were 96%, 91% and 82%, respectively. Survival in patients of WHO functional class II at baseline at 1, 2 and 3 years was 99%, 91%, and 84% respectively, and for patients of WHO functional class III at baseline was 94%, 90%, and 81%, respectively.
Efficacy in adult patients with PAH (when used in combination with epoprostenol): A randomized, double-blind, placebo controlled, study was conducted in 267 patients with PAH who were stabilized on intravenous epoprostenol. The PAH patients included those with Primary PAH, and PAH associated with CTD. Patients were randomized to placebo or sildenafil (in a fixed titration starting from 20 mg, to 40 mg and then 80 mg, three times a day) when used in combination with intravenous epoprostenol. The primary efficacy endpoint was the change from baseline at Week 16 in 6MWD. There was a statistically significant benefit of sildenafil compared to placebo in 6MWD. The mean change from baseline at Week 16 was 30.1 m for the sildenafil group compared with 4.1 m for the placebo group, giving an adjusted treatment difference of 26.0 m (95% CI: 10.8, 41.2) (p=0.0009). Patients on sildenafil achieved a statistically significant reduction in mean Pulmonary Arterial Pressure (mPAP) compared to those on placebo. A mean placebo-corrected treatment effect of -3.9 mmHg was observed in favor of sildenafil (95% CI: -5.7, -2.1) (p=0.00003).
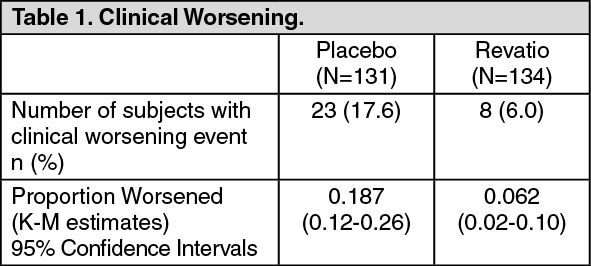
Delay in Clinical Worsening: Treatment with sildenafil significantly delayed the time to clinical worsening of PAH compared to placebo (p = 0.0074) with Kaplan-Meier (K-M) estimates demonstrating that placebo patients were 3 times more likely to experience an event (see Table 1). Time to clinical worsening was defined as the time from randomization to the first occurrence of a clinical worsening event (death, lung transplantation, initiation of bosentan therapy, or clinical deterioration requiring a change in epoprostenol therapy). 23 subjects experienced clinical worsening events in the placebo group (17.6%) compared with 8 subjects in the sildenafil group (6.0%). (See Table 1.)
 Click on icon to see table/diagram/image
Efficacy and safety in adult patients with PAH (when used in combination with bosentan):
Click on icon to see table/diagram/image
Efficacy and safety in adult patients with PAH (when used in combination with bosentan): A randomized, double-blind, placebo controlled study was conducted in 103 subjects with PAH who were on bosentan therapy for a minimum of three months. The PAH patients included those with primary PAH, and PAH associated with CTD. Patients were randomized to placebo or sildenafil (20 mg three times a day) in combination with bosentan (62.5-125 mg twice a day). The primary efficacy endpoint was the change from baseline at Week 12 in 6MWD. The results indicate that there is no significant difference in mean change from baseline on 6MWD observed between sildenafil 20 mg and placebo (13.62 m and 14.08 m, respectively).
Differences in 6MWD were observed between patients with primary PAH and PAH associated with CTD. For subjects with primary PAH (67 subjects), mean changes from baseline were 26.39 m and 11.84 m for the sildenafil and placebo groups, respectively. However, for subjects with PAH associated with CTD (36 subjects) mean changes from baseline were -18.32 m and 17.50 m for the sildenafil and placebo groups, respectively.
Overall, the adverse events were generally similar between the two treatment groups (sildenafil plus bosentan versus bosentan alone), and consistent with the known safety profile of sildenafil when used as monotherapy (see Dosage & Administration, Precautions and Interactions).
Effects on mortality in adults with PAH: A study to assess the effects of different dose levels of sildenafil on mortality in adults with PAH was conducted following the observation of a higher risk of mortality in pediatric patients taking a high dose of sildenafil three times a day, based on body weight, compared to those taking a lower dose in the long-term extension of the pediatric clinical trial (see Pediatric population and Long-term extension data as follows).
The study was a randomized, double-blind, parallel-group study in 385 adults with PAH. Patients were randomly assigned 1:1:1 to one of three treatment groups (5 mg three times a day (4 times lower than the recommended dose), 20 mg three times a day (recommended dose) and 80 mg three times a day (4 times the maximum recommended dose). In total, the majority of patients were PAH treatment naïve (83.4%). For most patients the etiology of PAH was idiopathic (71.7%). The most common WHO Functional Class was Class III (57.7% of patients). All three treatment groups were well balanced with respect to baseline demographics of strata history of PAH treatment and etiology of PAH, as well as the WHO Functional Class categories.
The mortality rates were 26.4% (n=34) for the 5 mg TID dose, 19.5% (n=25) for the 20 mg TID dose and 14.8% (n=19) with the 80 mg TID dose.
Pediatric population: A total of 234 subjects aged 1 to 17 years were treated in a randomized, double-blind, multi-center, placebo-controlled parallel-group, dose-ranging study. Subjects (38% male and 62% female) had a body weight ≥8 kg, and had primary pulmonary hypertension (PPH) [33%], or PAH secondary to congenital heart disease [systemic-to-pulmonary shunt 37%, surgical repair 30%]. In this trial, 63 of 234 (27%) patients were <7 years old (sildenafil low dose = 2; medium dose = 17; high dose = 28; placebo = 16) and 171 of 234 (73%) patients were 7 years or older (sildenafil low dose = 40; medium dose = 38; and high dose = 49; placebo = 44). Most subjects were WHO Functional Class I (75/234, 32%) or II (120/234, 51%) at baseline; fewer patients were Class III (35/234, 15%) or IV (1/234, 0.4%); for a few patients (3/234, 1.3%), the WHO Functional Class was unknown.
Patients were naïve for specific PAH therapy and the use of prostacyclin, prostacyclin analogues and endothelin receptor antagonists was not permitted in the study, and neither was arginine supplementation, nitrates, alpha-blockers and potent CYP450 3A4 inhibitors.
The primary objective of the study was to assess the efficacy of 16 weeks of chronic treatment with oral sildenafil in pediatric subjects to improve exercise capacity as measured by the Cardiopulmonary Exercise Test (CPET) in subjects who were developmentally able to perform the test, n = 115). Secondary endpoints included hemodynamic monitoring, symptom assessment, WHO functional class, change in background treatment, and quality of life measurements.
Patients were allocated to one of three sildenafil treatment groups (low, medium or high) or placebo. Actual doses administered were dependent on body weight (see Table 2).
Click on icon to see table/diagram/image
The proportion of subjects receiving supportive medicinal products at baseline (anticoagulants, digoxin, calcium channel blockers, diuretics and/or oxygen) was similar in the combined sildenafil treatment group (47.7%) and the placebo treatment group (41.7%).
The primary endpoint was a percentage change in VO
2peak from baseline to Week 16 assessed by CPET. Mean baseline peak volume of oxygen consumed (VO
2) values were comparable across the sildenafil treatment groups (17.37 to 18.03 mL/kg/min), and slightly higher for the placebo treatment group (20.02 mL/kg/min). See figure and Table 3. A total of 106 out of 234 (45%) subjects were evaluable for CPET, which comprised those children ≥ 7 years old and developmentally able to perform the test. Children <7 years (sildenafil combined dose = 47; placebo = 16) were evaluable only for the secondary endpoints.
Mean increases in VO
2peak percentage change from baseline at Week 16, were observed with all 3 sildenafil doses (range of 6.44% - 13.40%, figure), with little change with placebo (0.53%). The estimated difference between the combined sildenafil doses and placebo was 7.71% (95% CI: -0.19 to 15.60). See Table 3. (See figure and Table 3.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Estimates based on ANCOVA with adjustments for the covariates baseline VO
2peak, etiology and weight group.
Patients on sildenafil experienced dose dependent reductions in PVRI and mPAP compared to those on placebo (see Table 4).
Click on icon to see table/diagram/image
Significant improvements in functional class were demonstrated only in subjects on sildenafil high dose compared to placebo. Odds ratios for the sildenafil low, medium and high dose groups compared to placebo were 0.6 (95% CI: 0.18, 2.01), 2.25 (95% CI: 0.75, 6.69) and 4.52 (95% CI: 1.56, 13.10), respectively.
Long-term extension data: Of the 234 pediatric subjects treated in the short-term, placebo-controlled study, 220 subjects entered the long-term extension study. Subjects who had been in the placebo group in the short-term study were randomly reassigned to sildenafil treatment; subjects weighing ≤20 kg entered the medium or high dose groups (1:1), while subjects weighing >20 kg entered the low, medium or high dose groups (1:1:1). Of the total 229 subjects who received sildenafil, there were 55, 74, and 100 subjects in the low, medium and high dose groups, respectively. Across the short-term and long-term studies, the overall duration of treatment from start of double-blind for individual subjects ranged from 3 to 3129 days. By sildenafil treatment group, median duration of sildenafil treatment was 1696 days (excluding the 5 subjects who received placebo in double-blind and were not treated in the long-term extension study).
Peak VO
2 was assessed 1 year after the start of the placebo-controlled study. Of those sildenafil treated subjects developmentally able to perform the CPET 59/114 subjects (52%) had not shown any deterioration in Peak VO
2 from start of sildenafil. Similarly 191 of 229 subjects (83%) who had received sildenafil had either maintained or improved their WHO Functional Class at 1 year assessment.
Kaplan-Meier estimates of survival at 3 years in patients >20 kg in weight at baseline were 94%, 93% and 85% in the low, medium and high dose groups, respectively; for patients ≤20 kg in weight at baseline, the survival estimates were 94% and 93% for subjects in the medium and high dose groups respectively (see Precautions and Adverse Reactions).
Pharmacokinetics: Absorption: Sildenafil is rapidly absorbed. Maximum observed plasma concentrations are reached within 30 to 120 minutes (median 60 minutes) of oral dosing in the fasted state. The mean absolute oral bioavailability is 41% (range 25-63%). After oral three times a day dosing of sildenafil, AUC and C
max increase in proportion with dose over the dose range of 20-40 mg. After oral doses of 80 mg three times a day a more than dose proportional increase in sildenafil plasma levels has been observed.
When sildenafil is taken with food, the rate of absorption is reduced. In the presence of a high fat meal, there was a mean delay in T
max of 60 minutes and a mean reduction in C
max of 29%; however, the extent of absorption was not significantly affected (AUC decreased by 11%).
Bioequivalence was established between the 20 mg tablet and the 10 mg/mL oral suspension when administered as a 20 mg single oral dose of sildenafil (as citrate).
Distribution: The mean steady state volume of distribution (V
ss) for sildenafil is 105 L, indicating distribution into the tissues. After oral doses of 20 mg three times a day, the mean maximum total plasma concentration of sildenafil at steady state is approximately 113 ng/mL. Sildenafil and its major circulating N-desmethyl metabolite are approximately 96% bound to plasma proteins. Protein binding is independent of total drug concentrations.
Based upon measurements of sildenafil in semen of healthy volunteers 90 minutes after dosing, less than 0.0002% (average 188 ng) of the administered dose may appear in the semen of patients.
Metabolism: Sildenafil is cleared predominantly by the CYP3A4 (major route) and CYP2C9 (minor route) hepatic microsomal isoenzymes. The major circulating metabolite results from Ndemethylation of sildenafil. This metabolite has a phosphodiesterase selectivity profile similar to sildenafil and an
in vitro potency for PDE5 approximately 50% that of the parent drug. In healthy volunteers, plasma concentrations of this metabolite are approximately 40% of those seen for sildenafil. The N-desmethyl metabolite is further metabolized, with a terminal half-life of approximately 4 h. In patients with PAH, however, the ratio of UK- 103,320 to sildenafil is higher. Plasma concentrations of N-desmethyl metabolite are approximately 72% those of sildenafil after 20 mg three times a day dosing (translating into a 36% contribution to sildenafil's pharmacological effects). The subsequent effect on efficacy is unknown.
Elimination: The total body clearance of sildenafil is 41 l/h with a resultant terminal phase half-life of 3-5 h. After either oral or intravenous administration, sildenafil is excreted as metabolites predominantly in the faeces (approximately 80% of administered oral dose) and to a lesser extent in the urine (approximately 13% of administered oral dose).
Pharmacokinetics in special patient groups: Elderly: Healthy elderly volunteers (65 years or over) had a reduced clearance of sildenafil, resulting in approximately 90% higher plasma concentrations of sildenafil and the active N-desmethyl metabolite compared to those seen in healthy younger volunteers (18-45 years). Due to age-differences in plasma protein binding, the corresponding increase in free sildenafil plasma concentration was approximately 40%.
Renal impairment: In volunteers with mild to moderate renal impairment (creatinine clearance = 30-80 ml/min), the pharmacokinetics of sildenafil were not altered after receiving a 50 mg single oral dose. In volunteers with severe renal impairment (creatinine clearance < 30 ml/min), sildenafil clearance was reduced, resulting in mean increases in AUC and C
max of 100% and 88% respectively compared to age-matched volunteers with no renal impairment. In addition, N-desmethyl metabolite AUC and C
max values were significantly increased by 200% and 79% respectively in subjects with severe renal impairment compared to subjects with normal renal function.
Hepatic impairment: In volunteers with mild to moderate hepatic cirrhosis (Child-Pugh class A and B) sildenafil clearance was reduced, resulting in increases in AUC (85%) and C
max (47%) compared to age-matched volunteers with no hepatic impairment. The pharmacokinetics of sildenafil in patients with severely impaired hepatic function (Child Pugh class C) have not been studied.
Population pharmacokinetics: Age, gender, race, renal and hepatic function were included as factors in the population pharmacokinetic model to evaluate sildenafil pharmacokinetics in PAH patients. The data set available for the population pharmacokinetic evaluation contained a wide range of demographic data and laboratory parameters associated to hepatic and renal function.
None of the factors related to demographics, hepatic or renal function had a statistically significant impact on sildenafil pharmacokinetics in patients with PAH.
In patients with pulmonary arterial hypertension, the average steady state concentrations were 20-50% higher over the investigated dose range of 20-80 mg three times a day compared to healthy volunteers. There was a doubling of the C
min compared to healthy volunteers. Both findings suggest a lower clearance and/or a higher oral bioavailability of sildenafil in patients with pulmonary arterial hypertension compared to healthy volunteers.
Pediatric Patients: From a population PK model, body weight was shown to be a good predictor of drug exposure in children. Sildenafil plasma concentration half-life values were estimated to range from
2.9 to 4.4 hours for a range of 10 to 70 kg of body weight and did not show any differences that would appear as clinically relevant. The typical steady state C
max and AUC values following a 10 mg three times a day regimen to a 8 kg patient are expected to be 138 ng/mL and 1551 ng*hr/mL, respectively. The typical steady state C
max and AUC values following a 20 mg three times a day regimen to a 20 kg patient are expected to be 168 ng/mL and 1696 ng*hr/mL, respectively. T
max was estimated at approximately 1 hour and was almost independent from body weight.
Toxicology: Preclinical safety data: Preclinical data revealed no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, carcinogenicity potential, and toxicity to reproduction.
In pups of rats which were pre- and postnatally treated with 60 mg/kg sildenafil, a decreased litter size, a lower pup weight on day 1 and a decreased 4-day survival were seen at exposures which were approximately fifty times the expected human exposure at 20 mg three times a day. Effects in non-clinical studies were observed at exposures considered sufficiently in excess of the maximum human exposure indicating little relevance to clinical use.





 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image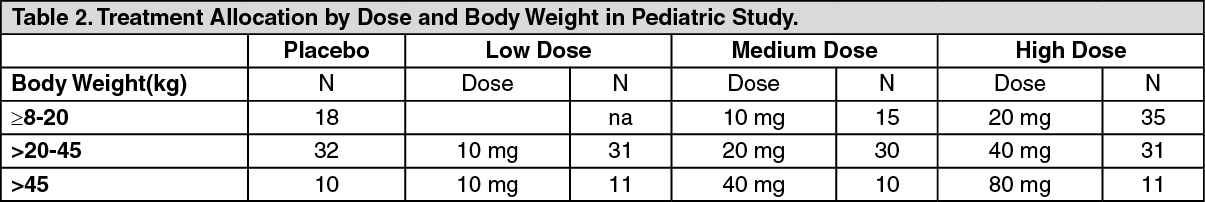 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image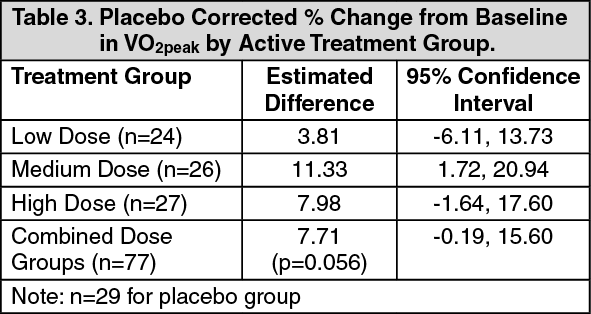 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
